{kind=link}
{kind=link}
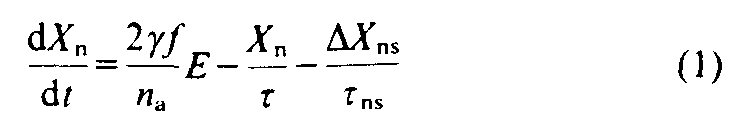
 Reproduced, with permission, from:
Elkins, J. W., T. M. Thompson, T. H. Swanson, J. H. Butler, B. D. Hall, S. O. Cummings, D. A. Fisher, and A. G. Raffo. 1993. Decrease in the growth rates of atmospheric chlorofluorocarbons 11 and 12. Nature 364: 780-83.
Reproduced, with permission, from:
Elkins, J. W., T. M. Thompson, T. H. Swanson, J. H. Butler, B. D. Hall, S. O. Cummings, D. A. Fisher, and A. G. Raffo. 1993. Decrease in the growth rates of atmospheric chlorofluorocarbons 11 and 12. Nature 364: 780-83.
J. H. Butler*, B. D. Hall*[[daggerdbl]], S. O. Cummings*+,
D. A. Fisher[[section]] & A. G. Raffo||
* National 0ceanic and Atmospheric Administration, Climate Monitoring and Diagnostics Laboratory, Boulder, Colorado 80303, USA
+ Cooperative Institute for Research in Environmental Sciences, University of Colorado, Boulder, Colorado 80309, USA
[[section]] E. I. DuPont de Nemours Co. & Inc., Scientific Computing Division, Central Research & Development, Wilmington, Delaware 19880, USA
|| E. I. DuPont de Nemours Co. & Inc., Fluorochemicals Division, Wilmington, Delaware 19898, USA
[[daggerdbl]] Current address: Washington State University, Department of Chemical Engineering, Pullman, Washington 99164, USA.
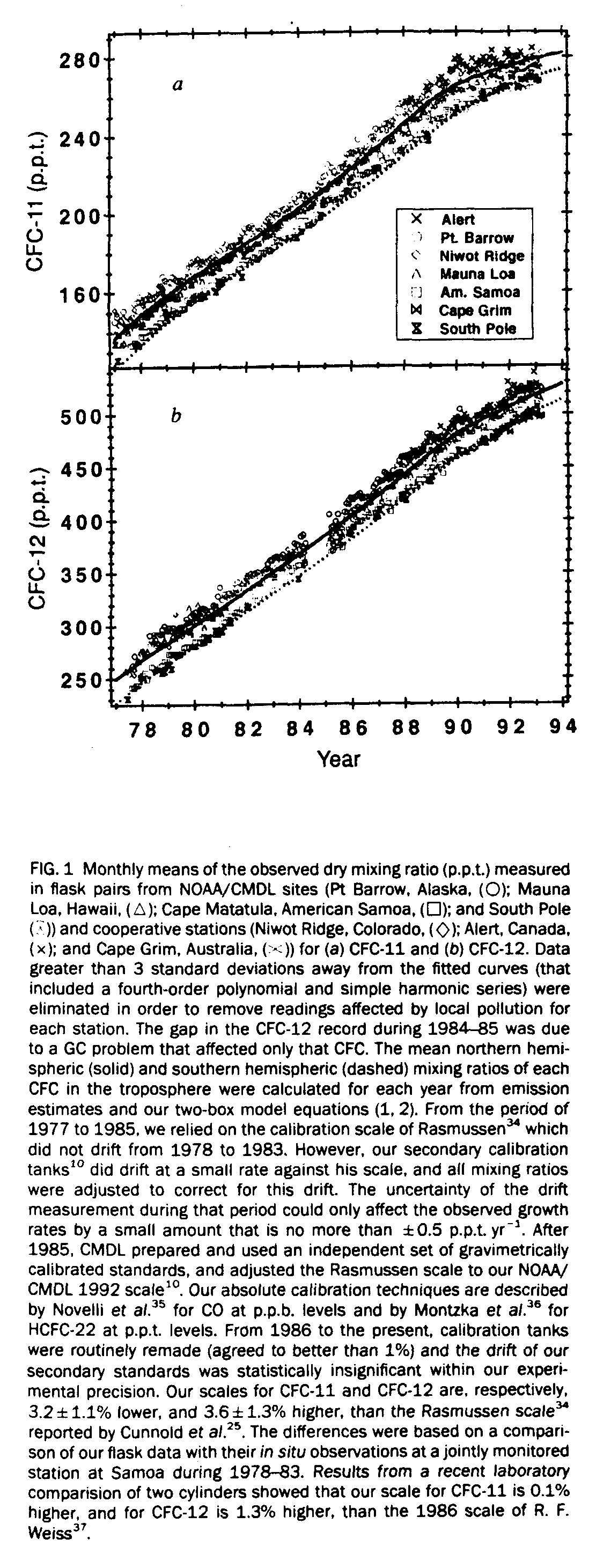
We report here the recent change in growth rates of tropospheric CFC-11 and -12, based on 15 years of data from flask samples and in situ instruments located at the monitoring stations of the National Oceanic and Atmospheric Administration's (NOAA) Climate Monitoring and Diagnostics Laboratory (CMDL). These stations are located at Pt Barrow, Alaska, 71.3[[ring]]N, 156.6[[ring]]W, elevation 8 m; Mauna Loa, Hawaii, 19.5deg.N, 155.6deg.W, 3,397m; Cape Matatula, American Samoa, 14.3[[ring]]S, 170.6[[ring]]W, 77 m, and South Pole, Antarctica, 90[[ring]]S, 2,841 m. Cooperative sampling sites were also employed, located at; Niwot Ridge, Colorado, 40.1[[ring]]N, 105.6[[ring]]W, 3,472 m; Alert, Canada, 82.5deg.N, 62.3[[ring]]W, 210 m, and Cape Grim Baseline Air Pollution Station. Australia, 40.7[[ring]]S, 144.8[[ring]]E, 94 m. Starting in 1977, paired samples were collected each week in electropolished, stainless steel flasks (with volumes of either 300 or 850 ml) at the four NOAA/CMDL stations and Niwot Ridge. After 1980, flask pairs at South Pole were collected once a week only during the austral summer when the station was open. Collection of flask pairs began at Alert in 1988, and at Cape Grim in 1991. All flasks were shipped back to Boulder for analysis by electron capture gas chromatography (EC-GC) [8, 9]. In 1987 our flask programme was complemented by automated in situ measurements [9, 10] (also performed by EC-GC) at the four NOAA/CMDL baseline observatories and Niwot Ridge. Measurements are reported as dry gas mole fractions or mixing ratios relative to the NOAA/CMDL 1991 gravimetric calibration scale [10].
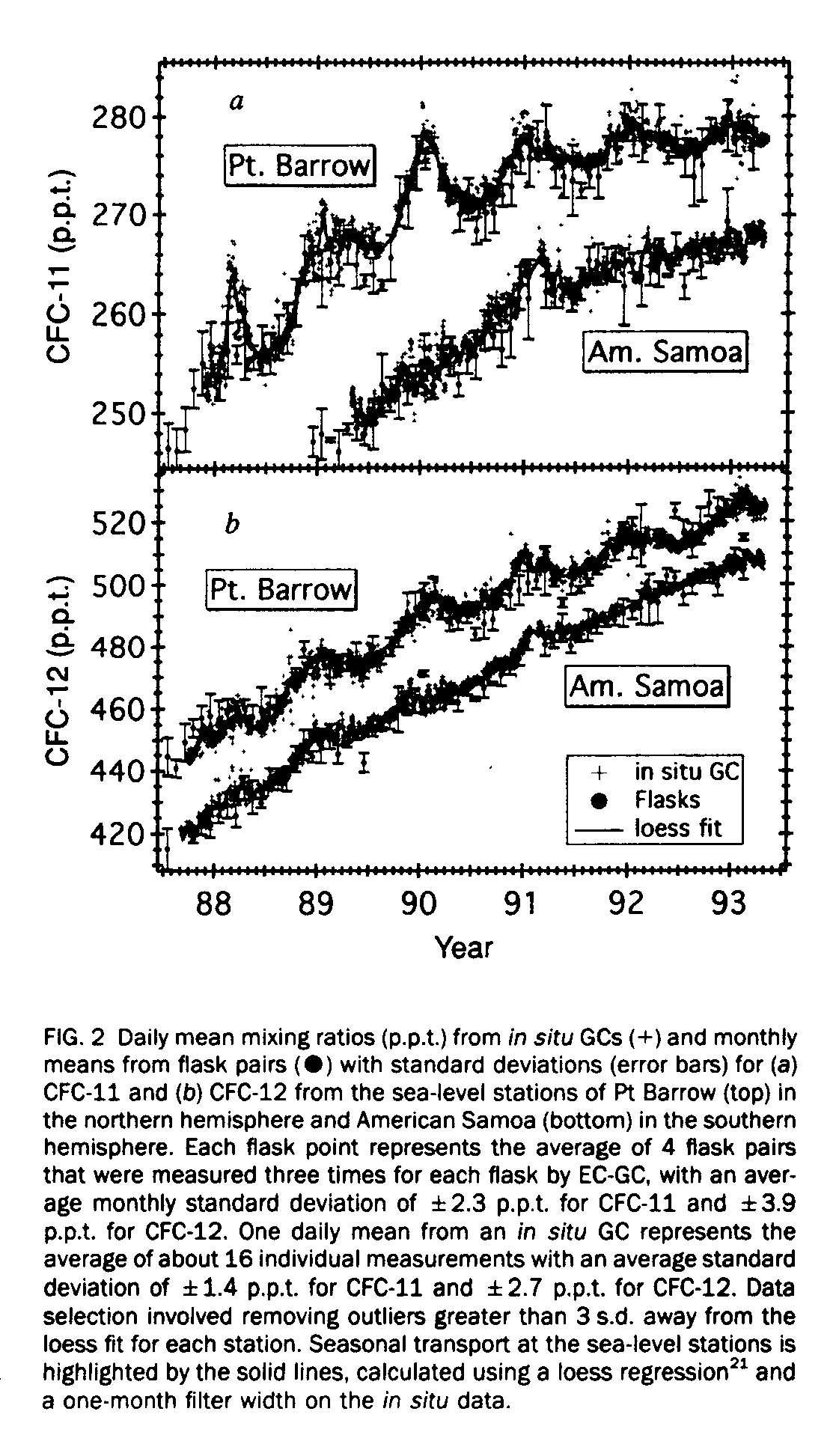
Figure 1 shows monthly means of the mixing ratios for atmospheric CFC-11 and -12 measured from more than 4,980 flask samples collected at NOAA/CMDL and cooperative stations between January 1977 and March 1993. The most obvious feature of the data is the levelling off of the mixing ratios, particularly CFC-11. Additional support for the slowdown of the atmospheric growth rates is presented in Fig. 2, which displays data collected (using in situ EC-GC measurements) at two sealevel sites, Pt Barrow and American Samoa. The seasonal patterns observed in the CFC data are primarily the result of transport. For Pt Barrow, the peak mixing ratios observed from November to March are the result of trapped cold air in the polar tropospheric vortex which restricts ventilation with cleaner air from the low latitudes". In contrast. the CFC maximum at American Samoa occurred during the period (November to March) of highest frequency of northwesterly flow which transports air masses enriched with CFCs from the northern hemispherere. It is important to note that the amplitude of the seasonal cycle for both CFCs has also decreased with time and that this decrease is consistent with reduced emissions.
The levelling off of mixing ratios after 1989 is a direct result of the reduced production of CFC-11 and -12 reported recently [7] by the CFC producers. DuPont scientists estimated the emissions [13] of both CFCs produced in the past, and those expected in the future, including the 1996 phaseout deadline. The procedures employed were published by Gamlen et al. [4] and are based on production surveys, estimates for production in non-surveyed countries, and market analyses for use after 1990. Using these new data for worldwide emissions and an approach similar to our one-box model [14], we calculated the annual mean mixing ratios of the CFCs for both hemispheres from 1930 to 2000 with a two-box. finite-increment model that describes the mean change in the tropospheric mixing ratio for a one-year interval. The differential equations for the mean annual change in the mixing ratios are for the northern hemisphere (n):
and for the southern hemisphere (s):
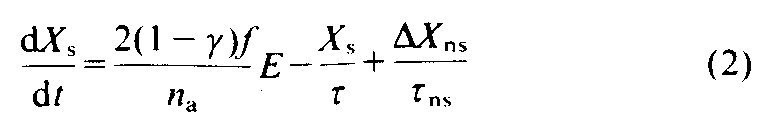
where X is the mean tropospheric mixing ratio in that hemisphere; is the ratio of the emissions in the northern hemisphere to the total emissions (~0.95); f is the fraction of total atmospheric CFC in the troposphere divided by the fraction of the total atmospheric mass of the troposphere [14], (because f has a latitudinal dependence, means that were weighted by the cosine of the latitude were calculated as 1.10+/-0 02 (one standard deviation, s.d.) for CFC-11 and 1.06+/-0.02 for CFC-12 from measurements taken aboard aircraft and balloons [15, 18], and spacecraft [19]); E is the emission rate (mol yr [-1]); na is the total mass of the atmosphere (moles);
is the mean atmospheric lifetime (yr);
ns is the mean interhemispheric exchange time [20] (1.1 yr), and
Xns is the mean interhemispheric difference. The best fits of the observed mixing ratios in Fig 1 were obtained with mean atmospheric lifetimes of 55 (+8, -9, 1 s.d.) yr for CFC-11 and 140 (+60, -35) yr for CFC-12. Thus using projected emissions [13] estimated by DuPont, the model predicts maximum mixing ratios for the northern hemisphere as ~290 parts per trillion, p.p.t. of CFC-11 in 1998 and ~555 p.p.t. of CFC-12 in 1999.
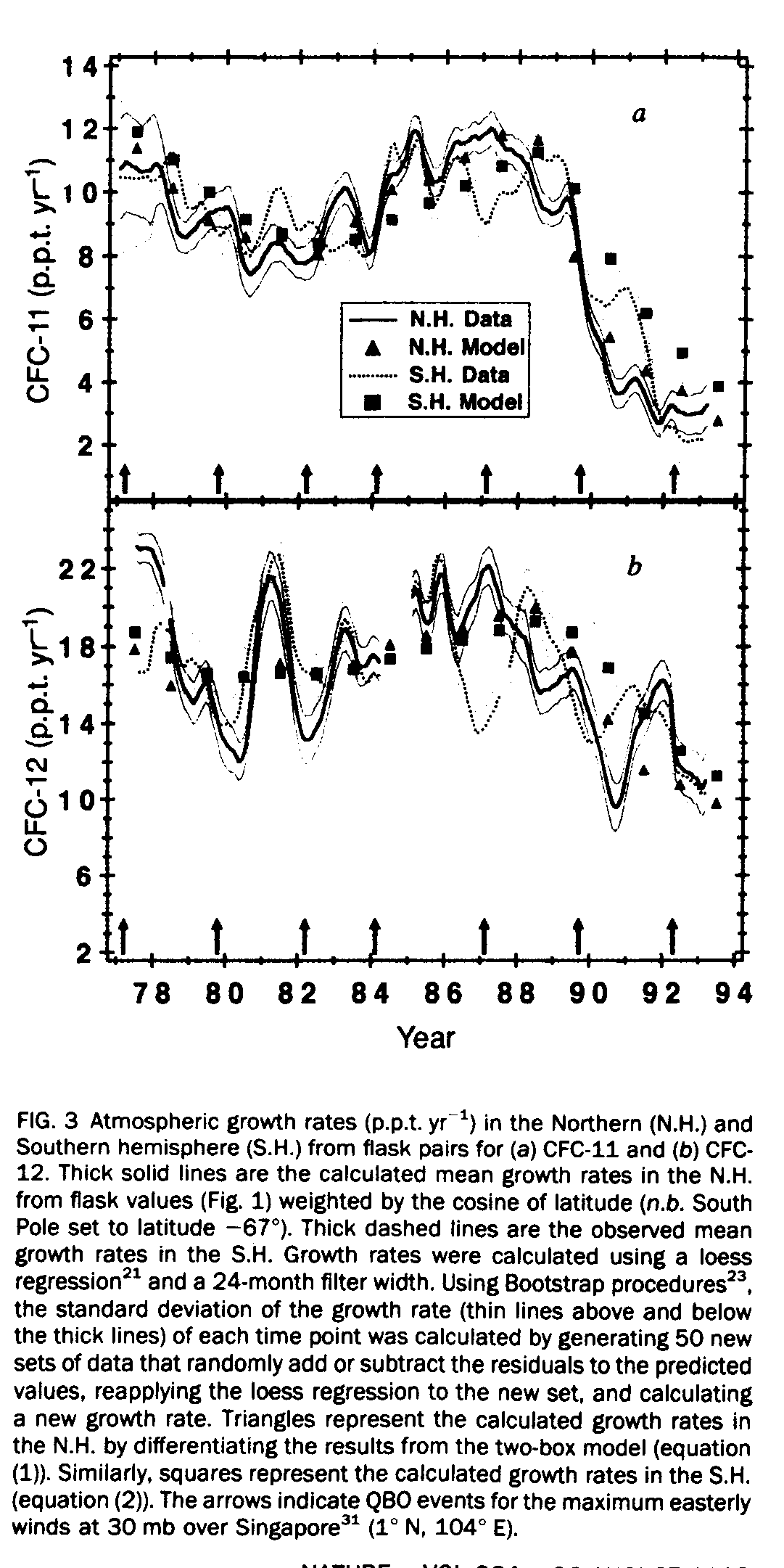
For further analysis of the growth rates for the CFCs, flask data were smoothed and filtered by the locally weighted least-squares (that is, loess regression [21]) procedure from the Dataplot graphics and statistical package [22]. The period of the data set that was locally weighted was fixed at 24 months, sufficient to examine the interannual variations of the growth rate. We estimated the growth rates by differentiating the time series of smoothed and filtered mixing ratios. These values were then compared to the ratio generated by our two-box model (Fig. 3). The uncertainty of the growth rates was estimated by applying the Bootstrap technique [23] to the residuals of the loess technique.
One of the most significant features of the observed data shown in Fig. 3 is the dramatic slowdown in growth rates for both CFCs beginning in late 1989. Previous reports [24, 25, 26] showed that the mean global growth rates between 1977 and 1984 were linearly increasing at a constant rate; our results give global rates of 9+/-1 (l.s.d.) p.p.t. yr[-1] for CFC-11 and 17+/-3 p.p.t. yr [-1] for CFC-12. Increased CFC usage during 1985-88 resulted in the observed growth rates climbing to average maxima for both hemispheres of 11+/-1 p.p.t.yr[-1] for CFC-11 and 19.5+/-2 p.p.t. yr[-1] for CFC-12. Global growth rates have decreased significantly since 1988, reaching levels of 2.7+/-0.7 p.p.t. yr[-1] for CFC-11 and 10.5+/-0.3 p.p.t. yr[-1] for CFC-12 by 1993. Perhaps the most significant fall-off of CFC-11 has occurred at the most northerly stations, Pt Barrow (Fig. 2) and Alert, where the rate of increase has essentially reached zero. This corresponds to the decrease in industrial production [6, 7] in the northern hemisphere where most of the material is used and therefore emitted. At the same time, growth in the southern hemisphere has decreased less rapidly because its growth rate is determined mainly by interhemispheric transport. The dramatic fall-off of CFC-11 is a result of its curtailed use particularly in the aerosol propellant and foam blowing applications in 1990. If these slowed growth rates are common for all compounds under regulation by the Montreal Protocol, the lower limit for the total cumulative organic chlorine, CCly, introduced into the stratosphere would be less than 4.0 parts per billion (p.p.b). (current value [5]= 3.4 +/- 0.2 p.p.b.).
Besides CFC production, other factors such as atmospheric transport may affect the observed hemispheric growth rates. A statistically significant divergence of the northern and southern hemispheric growth rates occurred during the period of the El Nino-Southern Oscillation (ENSO) event of 1986-87. This ENSO had a much stronger correlation with the growth rates of both CFCs than did the event of 1982-83. ENSO events, occurring every 4-7 years [27], are known to affect the interhemispheric transport of trace gases and have been correlated with lower than average mixing ratios of CH3CCl3 at Samoa [28]. Suppression of the westerly winds at 200 millibar (mb) in the upper troposphere which occurs during ENSOs [27] reduces the interhemispheric transport [29]. The variations observed in the hemispheric growth rates for both CFCs correlate well with hemispheric variations recently noted for methane [30] during 1986-89. A reason that the ENSO of 1986-87 had such a strong effect on the growth rates may be the coincidences of a quasi-biennial oscillation event (QBO), typically occurring every 26 months [29], followed by the strongest cold event in 15 years, the La Nina [27] of 1988-89. Also, there is a good correlation between the maximum of easterly winds of the equatorial stratosphere [31] (QBOs) and the drop in the southern hemispheric growth rate for both CFCs (Fig. 3). Although there is considerable evidence of tropospheric QBOs [32], dynamic models also show that strong easterly winds in the stratosphere can enhance vertical transport upward from the troposphere [33] and concurrently decrease the north to south tropospheric exchange [29]. As the mixing ratios of the CFCs have always been higher in the northern than southern hemisphere, any process that inhibits interhemispheric exchange or enhances tropospheric-stratosphere exchange will slow the growth rates in the southern hemisphere.
These decreased overall growth rates are encouraging in light of voluntary national and international efforts to limit emissions of ozone-depleting substances. Note that a recent report [14] also observed decreased atmospheric growth rates for the two dominant atmospheric halons, which are used primarily in fire extinguishers, and pose a significant threat to stratospheric ozone, although less than that posed by the CFCs considered here. Furthermore, continuous monitoring of CFCs, together with accurate tracking of emissions, will help to define the chemical lifetimes of these species and may provide a more complete understanding of the transport properties of the atmosphere.
1. Farman, J. C., Gardiner, B. G. & Shanklin, J. D. Nature 315, 207-210 (1985).
2. Molina, M. J. & Rowland, F. S. Nature 249, 810-814 (1974).
4. Gamlen, P. H., Lane. B. C., Midgley, P. M. & Steed, J. M. Atmos. Envir. 20(6), 1077-1085 (1986).
5. Prather, M. J. & Watson, R. T. Nature 344, 729-734 (1990).
6. McFarland, M. & Kaye, J. A. rev. J. Photochem Photobiol. 55, 911-929 (1992).
11. Barrie, L. A. & Hoff, R. M. Atmos. Envir. 13, 2711-2722 (1984),
12. Halter, B. C., Harris, J. M. & Conway, T. J. J. geophys Res 93(D12), 15914-15918 (1988).
16. Fabian, P., Borcher. R., Penkett, S. A. & Prosser N J D Nature 294, 733-735 (1981).
17. Fabian, P. et al. J. geophys. Res. 86(C6), 5179-5184 (1981).
18 Schmidt, U. et al. Geophys. Res Lett 18(4), 767-770 (1991).
19. Zander, R. et al. J. atmos. Chem. 15,171-186 (1992).
20. Jacob. D. J., Prather. M J.. Wofsy, S. C & McElroy, M B. J geophys Res 92(D2), 6614-6624 (1987).
21. Cleveland, W. S. J Amer. Stat. Assoc 74, 829-836 (1979).
22. Filliben, J. J. Comput. Graph. 15(3), 199-213 (1981).
23. Efron, B. & Tibshirani R. Science 253, 390-395 (1991).
24. Rasmussen, R. A. & Khalil, M. A. K. Science 232, 1623-1624 (1986).
25. Cunnold, D. M. et al. J. geophys. Res. 91(D10), 10797-10817 (1986).
28. Prinn, R. et al. J. geophys. Res. 97(D2), 2445-2461(1992).
29. Webster, P. J. & Holton, J. R. J. atmos. Sci. 39, 722-733 (1982).
30. Steele L. P. et al. Nature 360, 313-316 (1992).
32. Trenberth, K. E. Mon Weath. Rev. 108, 1370-1377 (1980).
33. Plumb, R. A. & Bell, R. C. Q. J. R. met. Soc. 100, 335-352 (1982).
34. Rasmussen, R. A. & Lovelock, J. E. J. geophys. Res. 88(C13), 8369-8376 (1983).
35. Novelli P. C.. Elkins J. W. & Steele, L. P. J. geophys. Res. 97(D7), 13109-13121(1991).
37. Bullister, J. L. & Weiss, R. F. Deep-Sea Res. 35(5), 839-853 (1988).
{kind=link}