 Reproduced, with permission, from:
Ravishankara, A. R., A. A. Turnipseed, N. R. Jensen, S. Barone, M. Mills, C. J. Howard, and S. Solomon. 1994. Do hydrofluorocarbons destroy stratospheric ozone? Science 263: 71-75.
Reproduced, with permission, from:
Ravishankara, A. R., A. A. Turnipseed, N. R. Jensen, S. Barone, M. Mills, C. J. Howard, and S. Solomon. 1994. Do hydrofluorocarbons destroy stratospheric ozone? Science 263: 71-75.
A. R. Ravishankara,* Andrew A. Turnipseed,+ Niels R. Jensen, Stephen Barone,* Michael Mills,+ Carleton J. Howard, Susan Solomon
National Oceanic and Atmospheric Administration, Aeronomy Laboratory, 325 Broadway, Boulder, CO 80303.
*Also affiliated with the Department of Chemistry and Biochemistry, University of Colorado, Boulder, CO 80309
+ CIRES, University of Colorado, Boulder, CO 80309.
Hydrofluorocarbons, many of which contain a CF3 group, are one of the major substitutes for the chlorofluorocarbons and halons that are being phased out because they contribute to stratospheric ozone depletion. It is critical to ensure that CF3 groups do not cause significant ozone depletion. The rate coefficients for the key reactions that determine the efficiency of the CF3 radical as a catalyst for ozone loss in the stratosphere have been measured and used in a model to calculate the possible depletion of ozone. From these results, it is concluded that the ozone depletion potentials related to the presence of the CF3 group in hydroflourocarbons are negligibly small.
The global phase-out of the ozone damaging chlorofluorocarbons (CFCs) and halons (chlorofluorobromocarbons) has led to various proposed substitutes. Many of the substitute compounds are hydrochlorofluorocarbons (HCFCs) and hydrofluorocarbons (HFCs). The short lifetimes of the HCFCs make them much less damaging to the ozone layer than the CFCs. The HFCs contain no chlorine or bromine, only fluorine. In contrast to chlorine and bromine, fluorine has been known to be benign toward the ozone layer. The HFCs have therefore been assumed to have ozone depletion potentials (ODPs) of essentially zero (1). (ODP is an index of the ozone depletion ability of a compound relative to CFC-11.) Under the U.S. Clean Air Act Amendments, only compounds with an ODP less than 0.2 may be used as halocarbon substitutes. The major HFCs under consideration or production are CF3CFH2 (134a), CF3CF2H (125), CHF3 (23), CF3CH3 (143a), and CF2HCH3 (152a). In particular, HCFC-134a has begun to be widely used as a substitute for CFCs in air conditioning and refrigeration applications. Many of these HFCs contain a CF3 functional group.
It has been recently suggested that molecules containing the CF3 group may represent a special case of fluorine-catalyzed ozone loss (2). The CF3 group is unusually stable and may destroy significant amounts of ozone through catalytic cycles involving CF3Ox (CF3O and CF3OO) radicals (2). If this were true, many HFCs (and possibly a few currently acceptable HCFCs) could have ODPs greater than 0.2, and new substitutes would have to be developed. The consequences of such a radical change in scientific understanding for industry, international agreements, and the phase-out of CFCs and HCFCs are far-reaching.
The important reactions of CF3O and CF3OO that may take place in the stratosphere are listed in Table 1. Reactions 1 and 2 constitute a catalytic cycle that is equivalent to 2O3 --> 3O2 There are many other possible catalytic cycles that are represented by the list of reactions, some of which are equivalent to 2O3 --> 3O2 and some to O + O3 --> 2O2.
All of the above catalytic cycles require that the CF3 group stay intact. If the CF3 group is degraded (as in reaction 3, for example), then the chain is terminated. In addition, reactions 4, 5, 8, and 11 lead to formation of CF3 reservoirs, which can also produce some ozone depletion (for example, through the cycle represented by reactions 1, 11, and 12 in conjunction with reaction of OH with O3). We have measured the rate coefficients for reactions 1 through 4, which are the key processes in these cycles, and used them to evaluate the ODPs of some of the most important HFCs that contain the CF3 group.
A pulsed photolysis apparatus equipped with pulsed laser-induced fluorescence detection (3) as well as a low-pressure flow tube coupled to a chemical ionization mass spectrometer (CIMS) detector (4) were used to study reactions 1 through 4. Use of diverse methods helps to identify possible systematic errors and is essential in resolving an issue as critical as the present one.
In the pulsed photolysis apparatus, CF3O radicals were generated by 193-nm laser photolysis of either bis-trifluoromethyl peroxide (CF3OOCF3) or CF3Br/O2/NO mixtures. CF3O was detected with laser-induced fluorescence by excitation of the A([2]A1) <-- X([2]E) electronic transition at 350.2 nm (0-0 band). The detection limit for CF3O (S/N = 1) was <7 x 10[9] molecules cm[-3] in 100 torr He upon averaging 100 laser shots. Typically, 10[11] to 10[12] molecules cm[-3] of CF3O were used. The delay time, that is, the reaction time, between the photolysis and the probe beam was varied between 10 us and 50 ms to allow construction of the temporal profile of CF3O.
In the flow tube apparatus, CF3OO radicals were produced in a side-arm reactor by pyrolysis of CF3I (producing CF3, which reacts with O2) and CF3O by pyrolysis of CF3OOCF3. CF3OO was reacted with I[-] to produce CF3O[-] or with SF6[-] to generate CF3OO[-]. CF3O was converted to CF3O[-] through its reaction with NO3[-] or SF6[-]. The ions were detected by a mass spectrometer. The reaction times were varied by moving an injector through which the stable reactants entered the flow tube. The detection limits for CF3O and CF3OO were ~10[9] molecules cm[-3]. Typically, ~5 x 10[10] molecules cm[-3] of CF3O and CF3OO were used.
All experiments were carried out in a large excess of pure He under pseudo-first order conditions in the concentration of free radical that was monitored. The concentrations of the excess reagent such as O3, NO, and CH4 were determined as described elsewhere (4, 5). CF3OOCF3 and O3 were measured by absorption at 184.9 nm and 254 nm (Hg lines), respectively.
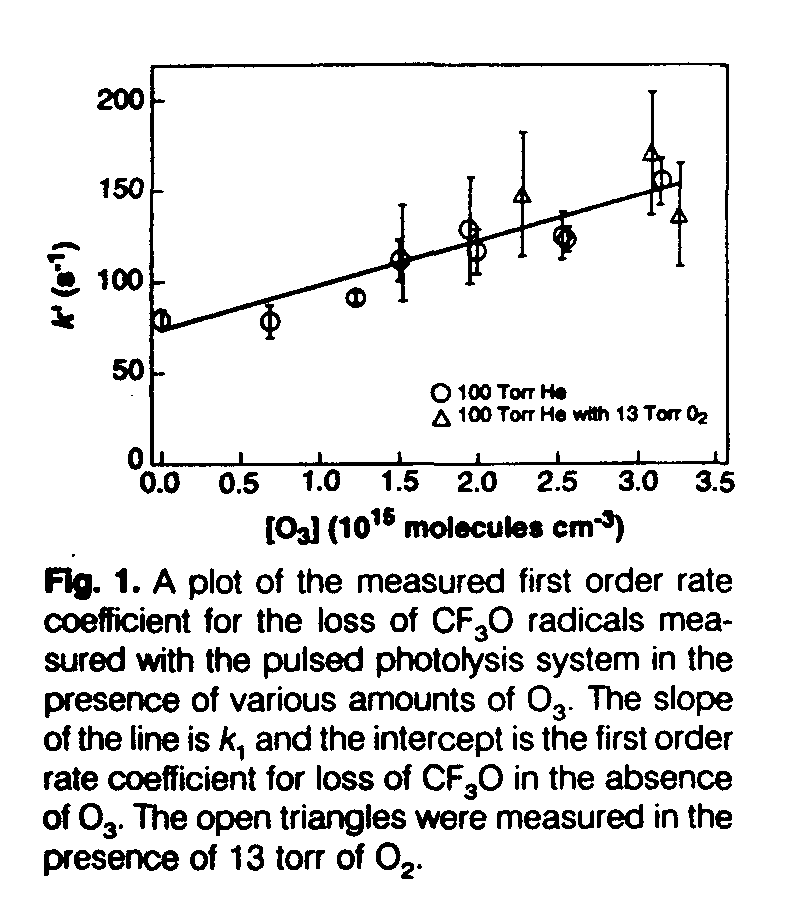
For reaction 1, the temporal profiles of CF3O in the presence of a large excess of O3 were observed to be exponential. From these profiles, the first order rate coefficient k1' = (k1[O3] + kd) was calculated. Here kd is the first order rate coefficient for the loss of CF3O in the absence of O3. Within the errors, k1' varied linearly with [O3] (Fig. 1). From these data, k1 = (2.5 +/- 1.0) x 10[-14] cm[3] molecule[-1] s[-1] at 298 K was determined in the pulsed photolysis system. The flow-tube study yields a consistent value (k1 < 4 x 10[-14] cm[3] molecule[-1] s[-1]). To be conservative in the ODP calculations, we used an upper limit of 4 x 10[-14] cm[3] molecule[-1] s[-1] for this rate coefficient (Table 1). Our other observations are consistent with the products of this reaction being CF3OO and O2 (see below). This reaction will likely be slower in the stratosphere, where the temperature will be much lower.
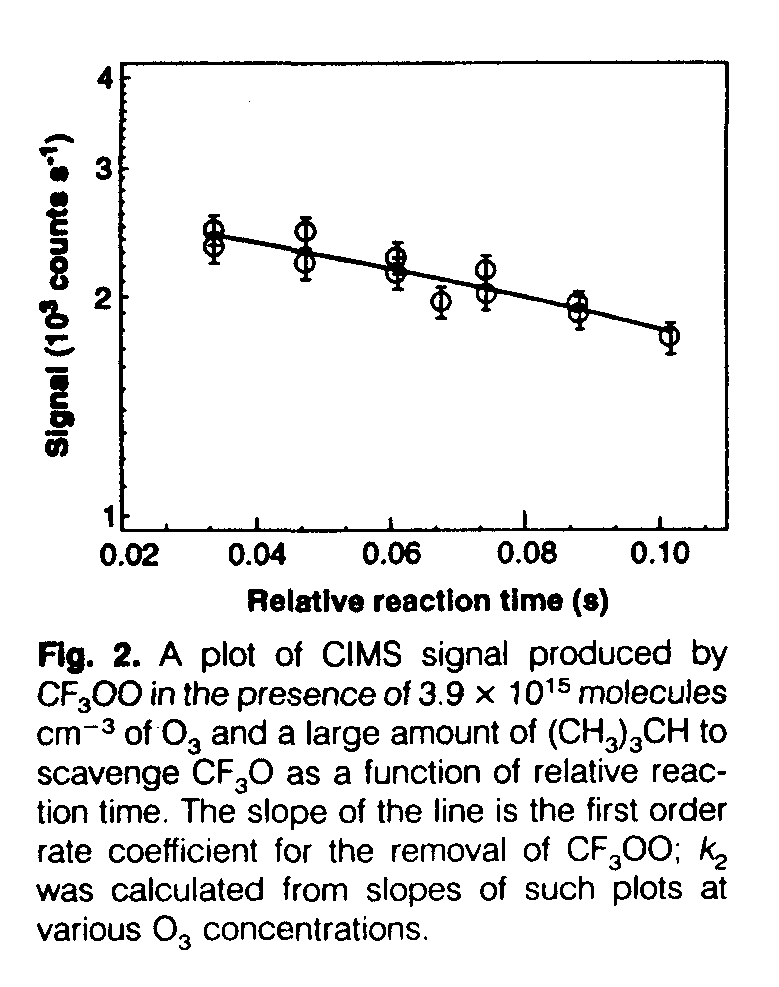
For reaction 2, the temporal profile of CF3OO was measured in the flow tube equipped with chemical ionization mass spectrometry (CIMS) detection in excess O3 (Fig. 2). As a means to inhibit the possible regeneration of CF3OO by the reaction of the product CF3O with O3, isobutane was added in great excess to scavenge the CF3O product through the reaction CF3O + (CH3)3CH --> CF3OH + (CH3)3C The loss of CF3) was very small in an excess of O3 (1.5 x 10[15] to 5.0 X 10[15] cm[-3]) when the isobutane scavenger was present; this indicates a very slow reaction, k2 < 3 x 10[-15] cm[3] molecule[-1] s[-1], if indeed any reaction does occur at all. In the absence of the CF3O scavenger, the temporal profile of CF3OO showed no loss, which suggests that if the slow reaction were occurring, it produces CF3O that in turn reacts with O3 to regenerate CF3OO. A low value of k2 is consistent with the low reactivity with O3 of other peroxy radicals such as HO2 (k = 2 x 10[-15] cm[3] molecule[-1] s[-1]) and CH3OO (k < 3 x 10[-17] cm3 molecule[-1] s[-1]) (6). An upper limit for k[2] at 298 K was also deduced from pulsed photolysis, where the ratio of k1/k2 was measured to be >4. On the basis of the quoted upper limit for k1, we calculate k2 < 1 x 10[-14] cm[3] molecule[-1] s[-1], consistent with the CIMS value. This rate coefficient is likely to be lower at stratospheric temperatures on the basis of analogy with the HO2 reaction.
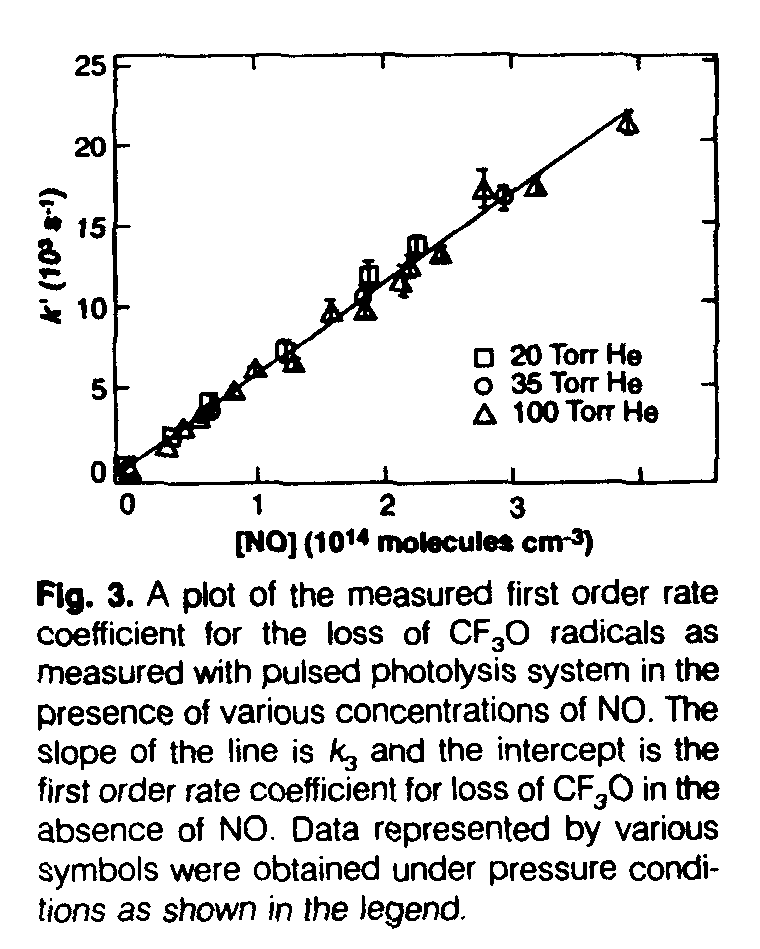
For reaction 3, the temporal profiles of CF3O measured in the photolysis system were strictly exponential and gave the values of the first order rate coefficient k3' = (k3[NO] + kd). In this expression, kd is the first order loss rate coefficient of CF3O in the absence of NO. A plot of k3' versus [NO] from one series of experiments is shown in Fig. 3. The slope of this line is k3 and yields a value of k3 = (5.45 +/- 0.57) x 10[-11] cm[3] molecule[-1] s[-1]. Similar experiments carried out in the flow tube yield k3 = (5.7 +/- 1.3) x 10[-11] cm[3] molecule[-1] s[-1], In the pulsed photolysis system, k3 and k13 were also measured by production of CF3O from CF3Br photolysis in the presence of NO and O2 to be (5.46 +/- 0.68) x 10[-11] and (1.57 +/- 0.38) x 10[-11] cm[3] molecule[-1] s-1, respectively. This value of k13 is in good agreement with previous determinations (4). The agreement between these three determinations of k3 is excellent and yields an average value of k3 = (5.6 +/- 0 7) x 10[-11] cm[3] molecule[-1] s[-1], independent of pressure. The products of this reaction have been shown to be CF2O + FNO (4, 7, 8). Preliminary measurements show that the rate coefficient for this radical-radical reaction increases slightly with decreases in temperature. Thus, the rate coefficients at stratospheric temperatures will be larger than the 298 K value (Table 1).
For reaction 4, k4 was also measured by following CF3O loss in the pulsed photolysis system as well as in the CIMS system in the presence of a large excess of CH4. The averaged 298 K value was (2.1 +/- 0.4) x 10[-14] cm[3] molecule[-1] s[-1], in good agreement with another recent measurement (9). Extrapolation of a few measured values of k4 at temperatures above 245 K to lower stratospheric temperatures of 200 K yields the lower limit shown in Table 1.
Estimates of the other rate coefficients needed to assess the role of CF3 in the stratosphere are given in Table 1. In the modeling calculations described below, it is assumed that CF3 is released instantaneously upon initiation of HFC degradation, primarily by OH or O([1]D) reactions, and converted into CF3OO in the atmosphere. All of the rate coefficients and photochemical parameters shown in Table 1 are either measured or estimated on the basis of analogies.
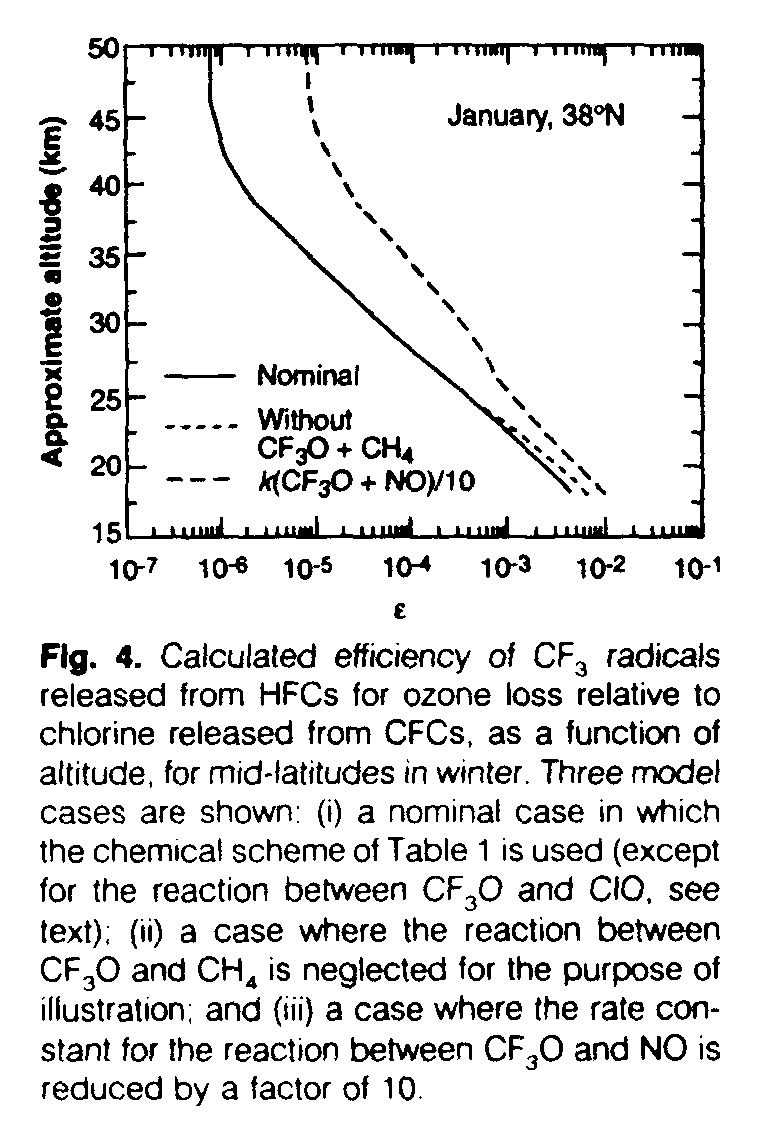
We evaluated the ODPs of several HFCs containing the CF3 group using both semiempirical and purely theoretical approaches (10, 11). The purely theoretical calculations were carried out with a two-dimensional (latitude and altitude) numerical model of the atmosphere (10) wherein perturbations of the HFC in question and a reference gas (CFC-11) are considered and the relative ozone loss is determined. The model includes heterogeneous chemistry on sulfate aerosols but not polar stratospheric clouds. All of the CF3-related species (see Table 1) were calculated from steady-state assumptions, and the chemistry of Table 1 was adopted with two important exceptions: (i) Reaction 8 (CF3O + ClO) was not considered because the present model does not include polar stratospheric cloud chemistry and related polar ozone depletion processes and (ii) the production of ozone by photolysis of NO2 formed from reaction 13 was neglected to maximize the possible ozone loss and, hence, present a worst case. Sensitivity tests were also carried out to ensure that reaction 8 is not significant as compared with others that were included and, therefore, not a serious shortcoming. For the semiempirical calculations, the distribution of the HFC source gas 134a was inferred from an observationally derived CH4 distribution in which the correlation of HFC-134a with CH4 determined from the two-dimensional model was used. The relative efficiency of CF3 released from HFC-134a for ozone loss as compared with chlorine released from CFCs was evaluated in the two-dimensional model and used as input to the semiempirical calculation. Figure 4 shows the value of this important parameter (hereafter referred to as E), illustrating the fact that the CF3 radicals are most efficient for ozone loss relative to chlorine in the lowest part of the stratosphere. This result might be expected from the fact that CF3O and CF3OO are chemically similar to OH and HO2.
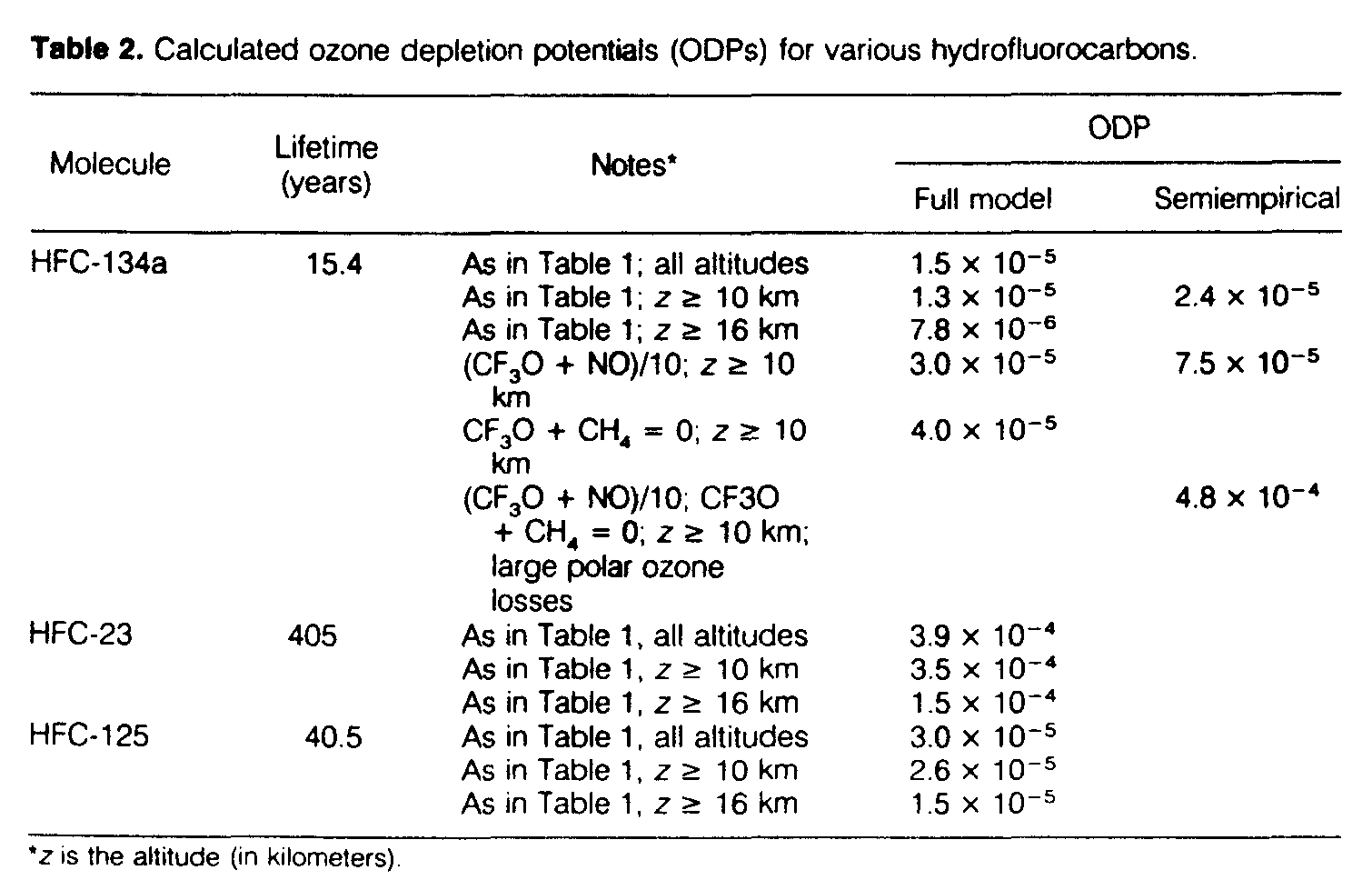
Table 2 presents the ODPs calculated for HFC-134a, -23, and -125. The lifetimes of these three species are between about 15 and 400 years, bracketing the range for the important HFCs. For the semiempirical calculations, a range of possible ozone loss profiles based on measurements was adopted as described in Solomon et al. (11). The table shows the highest values derived as an upper limit, which were obtained when no vortex processing was considered and maximum ozone changes were adopted at the lowest altitudes; see Solomon et al. (11). The semiempirical values are similar to those of the complete model but tend to be somewhat larger, owing mainly to the fact that the theoretical model tends to underestimate the observed ozone losses in the lowest part of the stratosphere. The semiempirical calculation includes only observed ozone losses above ~12 km; as Table 2 indicates, losses at lower altitudes do play a role in the theoretical model calculations. The calculated ODPs for HFC-125 and HFC-23 are greater than that of HFC-134a because of their longer lifetimes, but are still found to be well below 1 x 10[-3].
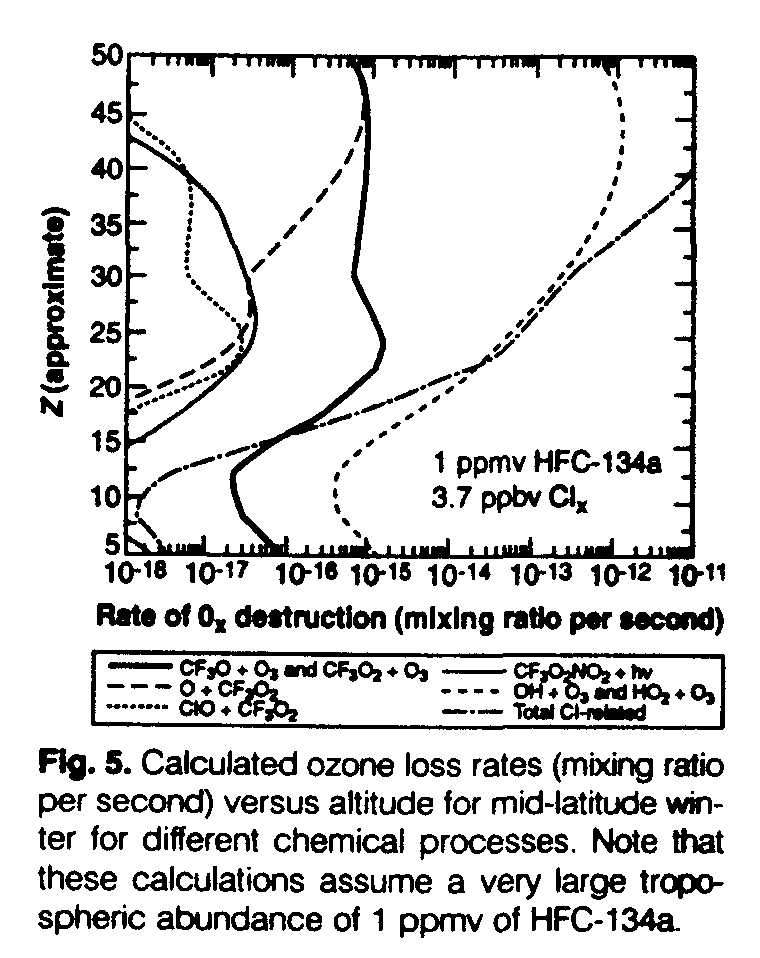
We next present a calculation showing the expected benefit to the ozone layer of substitution of HFC-134a for current CFC use. The calculation is intended to be illustrative rather than realistic. A comparison of the calculated ozone loss rates resulting from different chemical processes for mid-latitudes (Fig. 5) shows that the shape of the ozone loss profile related to CF3 chemistry is similar to that of OH + O3 and HO2 + O3 and is much steeper than that of chlorine-related chemistry (compare with Fig. 4). Figure 5 shows that the ozone depletion related to the reservoirs considered (CF3OOH and CF3OONO2) is negligible. Indeed, if these reservoirs are short-lived (that is, an instantaneous balance between production and loss can be assumed in the stratosphere) and their ozone destruction can be neglected, then they play no role in determining the ODP. To obtain a globally averaged ozone loss of 1% that is illustrated in Fig. 5, an HFC-134a tropospheric abundance of 1 ppmv was needed. Such a large, albeit unrealistic, abundance was used to ensure the accuracy of the calculated O3 change and, more importantly, to show that a very large total global release of HFC-134a of about 1.5 billion metric tons per year would be required to produce a globally averaged ozone loss of ~1%. This number can be compared to the global use of all CFCs, estimated at about 700,000 metric tons in 1991. This calculation therefore implies that even in the case of complete substitution of HFC134a for all CFC use, ozone depletion would be expected to be markedly reduced.
In addition to the O3 loss mechanisms and CF3Ox reactions that we have explicitly considered, there are other proposed cycles and reactions that may affect the chemistry of CF3Ox in the stratosphere. The cycle involving the reaction of O with CF3O, proposed by Li and Francisco (12), is one such scheme. Our measured value of k3 is close to that possible for the reaction of CF3O with O, and the abundance of NO will exceed that of O atoms below ~40 km. Further, if the products of the CF3O + O reaction are CF2O + FO, this reaction will terminate, rather than propagate, the catalytic chain. Therefore, this reaction scheme was not included here. Similarly, we have neglected the reaction of CF3O with O2, which will lower the calculated efficiency of CF3 in destroying O3 because it will generate CF2O. The chemistry of FNO has not been included, because it absorbs quite strongly in the 290 to 340 nm region (13) and, hence, will photolyze rapidly to F and NO. The F atoms will be quickly converted to HF through reactions with CH4, H2O, and so forth in the stratosphere.
The calculations presented here show that the ozone depletion caused by the HFCs should be greatest in the lower stratosphere. The ODP depends primarily on the rate constant for the reaction of CF3O with O3 (which causes ozone loss) as compared to the reactions of CF3O with NO and CH4 (which terminate the catalytic cycle; Fig. 4 illustrates that both are important in the lower stratosphere). Thus, the results presented here depend to some extent on the modeled NO distribution. An extreme sensitivity test was therefore carried out in which the rate constant for the reaction of CF3O with NO (or, alternatively, the NO concentration) was decreased by a factor of 10. As can be seen from Table 2, even in this case the calculated ODP of HFC-134a for both the semiempirical and theoretical models is less than 1 x 10[-4]. Similarly, in the extreme case of neglecting completely the reaction of CF3O with CH4 (or, equivalently, assuming that the product CF3OH is short-lived and regenerates CF3), an ODP of about 4 x 10[-5] is estimated for HFC-134a from the theoretical model. The most extreme case considered was one in which the reaction of CF3O with CH4 was neglected, the reaction of CF3O with NO was divided by a factor of 10, and large polar ozone losses were included in the semiempirical model. Even for this very unrealistic simulation, the estimated ODP for HFC-134a was less than 5 x 10[-4], largely because the rate of formation of CF3 is slow in the high-latitude lower stratosphere and because chlorine chemistry is quite effective at destroying ozone there.
The kinetics measurements described here together with modeling and semiempirical estimates show that the chemical reactions involving CF3O and CF3OO lead to negligibly small ODPs. Therefore, we conclude that HFCs and HCFCs containing CF3 groups are no more harmful to the stratospheric ozone layer than those that do not. For example, the ODP for HCFC-123 will be determined by its chlorine release, with the presence of the CF3 group making essentially no contribution to the total. On the basis of our current understanding, it appears highly likely that the ODPs for the HFCs considered here are all well below 1 x 10[-3]. For the key substitute HFC-134a, the best estimate of the ODP is only 1 x 10[-5] to 2 x 10[-5].
Note added in proof: Since the submission of this manuscript, several studies on reaction 1 (14-6), reaction 2 (14, 15), reaction 3 (17), and reaction 13 (17) have been published. All of these measurements are in agreement with the results reported here.
3. A. A. Turnipseed, S. B Barone, A. R. Ravishankara, J. Phys. Chem. 96, 7502 (1992).
4. T. B Bevilacqua, D. R. Hanson, C. J. Howard, ibid 97, 3705 (1993).
5. A. A. Turnipseed, S. B. Barone, A. R. Ravishankara, ibid, p. 5926.
7. Z. Li and J. S. Francisco, Chem. Phys. Lett. 186, 336 (1991).
8. J. Chen, T. Zhu, H. Niki, J. Phys. Chem. 96, 6615 (1992).
9. H. Saathoff and R. Zellner, Chem. Phys. Lett. 206, 349 (1993).
10. R. R. Garcia, F. Stordal, S. Solomon, J. T. Kiehl, J Geophys. Res. 97, 12967 (1992).
11. S. Solomon et al, ibid., p. 825.
13. H. S. Johnston and H. J Bertin, J. Mol. Spectrosc. 3, 683 (1959)
14. O. J. Nielsen and S. Sehested. Chem. Phys. Lett. 213, 433 (1993)
15. M. Maricq and J. J. Szente, ibid, p. 449.
16. T. J. Wallington, M. D. Hurley, W. F. Schneider, ibid., p. 442.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}